

We are pleased to announce that the next CCG North American UGM and Conference will take place June 25-28 in Montreal, Canada. This is a 4-day event, consisting of workshops, scientific presentations, and a poster session as well as social activities including receptions and a conference dinner. 2024 marks Chemical Computing Group's 30th anniversary. Join us to celebrate such a special occasion!
If you would be interested in presenting at the meeting, please contact Raul Alvarez at .
Registration Opens (Check-in/Badge pick-up)
Welcome Refreshments
Ligand-Based Drug Design and SAR Analysis
MOEsaic / Matched Molecular Pairs (MMP) Analysis / R-Group Profiles and Analysis / Similarity and Substructure Searching / Molecular Property Analysis / Conformational Searching / Molecular Alignments / Pharmacophore Modeling and Searching
MOEsaic / Matched Molecular Pairs (MMP) Analysis / R-Group Profiles and Analysis / Similarity and Substructure Searching / Molecular Property Analysis / Conformational Searching / Molecular Alignments / Pharmacophore Modeling and Searching
Peptide Modeling, Conformational Searching and Docking
Peptide Complex Preparation / Protein-Peptide Interaction Analysis / Surfaces and Maps / Peptide Sequence Optimization / Non-Natural Amino Acids / Conformational Searching / Peptide-Protein Docking / Protein-Peptide Interaction Fingerprints
Peptide Complex Preparation / Protein-Peptide Interaction Analysis / Surfaces and Maps / Peptide Sequence Optimization / Non-Natural Amino Acids / Conformational Searching / Peptide-Protein Docking / Protein-Peptide Interaction Fingerprints
Introduction to SVL
SVL / Programming Language / Create Scripts / Customize MOE
SVL / Programming Language / Create Scripts / Customize MOE
Lunch Break
Small Molecule Virtual Screening
Virtual Screening Compound Libraries / Molecular Descriptors / QSAR/QSPR Modeling / Molecular Fingerprints / Pharmacophore Modeling / Filtering Compound Libraries / Pharmacophore-guided Docking / Template-based Docking / De novo Hit Expansion
Virtual Screening Compound Libraries / Molecular Descriptors / QSAR/QSPR Modeling / Molecular Fingerprints / Pharmacophore Modeling / Filtering Compound Libraries / Pharmacophore-guided Docking / Template-based Docking / De novo Hit Expansion
Antibody Modeling and Protein Engineering
Protein-Protein Interaction Analysis / Molecular Surfaces / Protein Patch Analysis / Protein Properties / Protein Engineering / Antibody Homology Modeling / Antibody Database / Developability Analysis
Protein-Protein Interaction Analysis / Molecular Surfaces / Protein Patch Analysis / Protein Properties / Protein Engineering / Antibody Homology Modeling / Antibody Database / Developability Analysis
Cheminformatics: Manage, Analyze, Model and Mine Molecular Data
MOE databases / Molecular Descriptors / Sorting and Coloring Plots / Clustering | Diverse Subset Selection / QSAR Modeling / Binary QSAR / Substructure Searching / Molecular Fingerprints / Similarity Searching
MOE databases / Molecular Descriptors / Sorting and Coloring Plots / Clustering | Diverse Subset Selection / QSAR Modeling / Binary QSAR / Substructure Searching / Molecular Fingerprints / Similarity Searching
Welcome Refreshments
Advanced Structure-Based Design
Protein-Ligand Interaction Analysis / Pharmacophore Modeling / Docking / Protein-Ligand Interaction Fingerprints (PLIF) / Fragment-based Design / Scaffold Replacement
Protein-Ligand Interaction Analysis / Pharmacophore Modeling / Docking / Protein-Ligand Interaction Fingerprints (PLIF) / Fragment-based Design / Scaffold Replacement
Biologics: Protein Alignments, Modeling and Docking
Protein Alignments and Superposition / Loop and Linker Modeling / Homology Modeling / Protein-Protein Docking / Epitope Analysis / Protein Properties / Protein Solubility Prediction / Protein Patches (2D and 3D) / Biologics QSAR/QSPR Modeling
Protein Alignments and Superposition / Loop and Linker Modeling / Homology Modeling / Protein-Protein Docking / Epitope Analysis / Protein Properties / Protein Solubility Prediction / Protein Patches (2D and 3D) / Biologics QSAR/QSPR Modeling
Fragment-Based Drug Design: Scaffold Replacement, Fragment Linking, R-Group Exploration and Bioisosteric Replacements
Scaffold Hopping / Fragment Linking / Ligand Growing / R-Group Screening / Bioisosteric Transformations / Pharmacophore Modeling / Fragment Databases
Scaffold Hopping / Fragment Linking / Ligand Growing / R-Group Screening / Bioisosteric Transformations / Pharmacophore Modeling / Fragment Databases
Lunch Break
Opening Remarks
Adventures in Large Chemical Space
Hans Purkey, Senior Director & Senior Principal Scientist, Computational Drug Discovery,Genentech
Hans Purkey, Senior Director & Senior Principal Scientist, Computational Drug Discovery,
Machine Learning Guided AQFEP: A Fast Efficient Absolute Free Energy Perturbation Solution for Virtual Screening
Andrea Bortolato, Director of Drug Discovery,SandboxAQ
Andrea Bortolato, Director of Drug Discovery,
Proteins, Partners, Pockets: Drug Discovery Exploration in an Integrated Computational-Experimental Structural Ecosystem
Seth F. Harris, Director Computational Structural Biology & Distinguished Scientist,Genentech
Seth F. Harris, Director Computational Structural Biology & Distinguished Scientist,
Afternoon Break
Advancing Force Field Design and Development at Open Force Field
Lily Wang, Science Lead,Open Force Field Initiative
Lily Wang, Science Lead,
AmberEHT Forcefield: Novel Approaches to Small Molecule Parameterization
Paul Labute, President and CEO,Chemical Computing Group
Paul Labute, President and CEO,
Opening Reception & Poster Session
Group Activities (Choice of Montreal Ferris Wheel or Brewery Beer Tasting)
Welcome Refreshments
From Water Structure to Pharmacophore Search
Dan McKay, Executive Director,Ventus Therapeutics
Dan McKay, Executive Director,
The Art of Designing Out: Using Structure and Ligand Based Designing Principles to Prioritize Safer Chemotype
Arijit Basu, Principal Scientist,Takeda
Arijit Basu, Principal Scientist,
MOE Deployment at Gilead & Unraveling of Cryptic Pockets
Wilian Cortopassi, Senior Research Scientist II,Gilead Sciences
Wilian Cortopassi, Senior Research Scientist II,
Morning Break
Historic Walking Tour
Lunch Break
Using Generative AI to Design Mini-Therapeutic Proteins Targeting a Heterodimeric Complex without a Solved Structure
Lindsay Hammack, Senior Scientist,Merck
Lindsay Hammack, Senior Scientist,
Tuning Coarse-grained, Monte Carlo Simulations to Screen for mAb Colloidal Stability
Joel Janke, Scientist, Biologic Drug Product Development & Manufacturing,Sanofi
Joel Janke, Scientist, Biologic Drug Product Development & Manufacturing,
Antibody Sequence and Structure-Based Workflows for Diversity and Developability Assessments
Vinodh Kurella, Senior Scientist,Takeda
Vinodh Kurella, Senior Scientist,
Afternoon Break
Dynamic Epitope Analysis of Antiviral mAbs
Kevin Hauser, Senior Scientist I, Computational Structural Biology,Vir Biotechnology, Inc.
Kevin Hauser, Senior Scientist I, Computational Structural Biology,
Computational Models to Predict and Guide Mitigation of Chemical Degradation in Antibodies
Saeed Izadi, Senior Principal Scientist & Group Leader,Genentech
Saeed Izadi, Senior Principal Scientist & Group Leader,
A Pharmacophore-Based Virtual Screening Method for Identifying Potential Antibody: Antigen Binding Partners
Sandeep Kumar, Distinguished Fellow (Executive Director), Head of Computational Protein Design and Modeling,Moderna Inc.
Sandeep Kumar, Distinguished Fellow (Executive Director), Head of Computational Protein Design and Modeling,
Conference Reception and Dinner Cruise
Welcome Refreshments
Beyond Ro5 Free Ligand Conformational and Dynamical Determinants of Passive Permeability: Computational and Experimental Synergies
Amber Y.S. Balazs, Director, US Analytical, Structural, and Chromatography (ASC) Team, Early Oncology Chemistry NMR Specialist,AstraZeneca
Amber Y.S. Balazs, Director, US Analytical, Structural, and Chromatography (ASC) Team, Early Oncology Chemistry NMR Specialist,
Understanding the Chameleonicity and Permeability of PROTACs Using Molecular Dynamics Simulations, Markov Models, and Deep Learning
Bryn Taylor, Scientist, In Silico Discovery,Johnson & Johnson Innovative Medicine
Bryn Taylor, Scientist, In Silico Discovery,
Morning Break
Platform To Enable Structure-Enriched Models For Challenging Systems: Strengthening Rational Design via Ternary Complex Models Triaged Based on Hydrogen-Deuterium Exchange Data (HDX)
Usha Viswanathan, Scientist,Johnson & Johnson Innovative Medicine
Usha Viswanathan, Scientist,
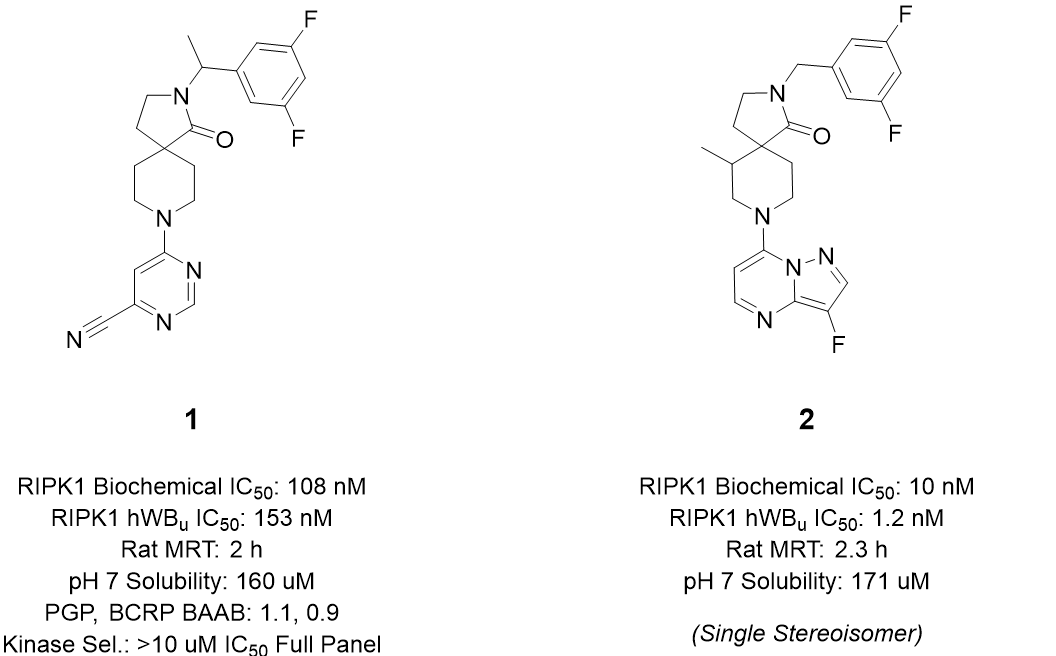
Discovery of Potent, Selective Type III RKIPK1 Inhibitors for the Treatment of Neurodegenerative Disease via SBDD and Library Design
Zach Brill, Associate Principal Scientist,Merck
Zach Brill, Associate Principal Scientist,
José Duca, Global Head of Computer-Aided Drug Discovery,
Closing Remarks
Lunch Break
Closing Desserts & Drinks Reception