

Morning Coffee
Registration (check-in/badge pick-up)
Registration (check-in/badge pick-up)
Analyzing Antibody-Antigen Complexes and Property Predictions
Opening, preparing & annotating protein complexes / Calculating protein properties & Patch Analyzer application / Analyzing protein contacts at Fab-antigen interface / Molecular surface & maps / Virtual mutagenesis with Protein Builder & Design
Opening, preparing & annotating protein complexes / Calculating protein properties & Patch Analyzer application / Analyzing protein contacts at Fab-antigen interface / Molecular surface & maps / Virtual mutagenesis with Protein Builder & Design
Morning Break
Antibody Homology Modeling and Structural Bioinformatics
Template & loop searching with Antibody Modeler / Building homology models of the Fv domain / Identifying & removing glycosylation sites / Antibody Database Project Search panel – Viewing antibody structure statistics / Building humanized Fab models
Template & loop searching with Antibody Modeler / Building homology models of the Fv domain / Identifying & removing glycosylation sites / Antibody Database Project Search panel – Viewing antibody structure statistics / Building humanized Fab models
Workshop Lunch
Registration (check-in/badge pick-up)
Opening Remarks
Discovery and Multimerization of Cross-Reactive Single-Domain Antibodies Against Sars-Like Viruses to Enhance Potency and Address Emerging SARS-CoV-2 Variants
Kalyan Pande, Principal Scientist, Discovery Biologics,Merck Research Labs
Cameron Noland , Associate Principal Scientist, Structural Chemistry, Merck Research Labs
Kalyan Pande, Principal Scientist, Discovery Biologics,
Cameron Noland
Deciphering Deamidation and Isomerization in Therapeutic Proteins: Effect of Neighboring Residue
Saeed Izadi, Senior Principal Scientist & Group Leader,Genentech
Saeed Izadi, Senior Principal Scientist & Group Leader,
A pan-influenza antibody inhibiting neuraminidase via receptor mimicry: How molecular dynamics-enabled epitope analysis helps explain activity
Kevin Hauser, Senior Scientist I, Computational Structural Biology,Vir Biotechnology, Inc.
Kevin Hauser, Senior Scientist I, Computational Structural Biology,
Structure-Based Charge Calculations for Predicting Properties and Profiling Antibody Therapeutics
Philippe Archambault, Applications Scientist,Chemical Computing Group
Philippe Archambault, Applications Scientist,
Afternoon Break - Poster Viewing
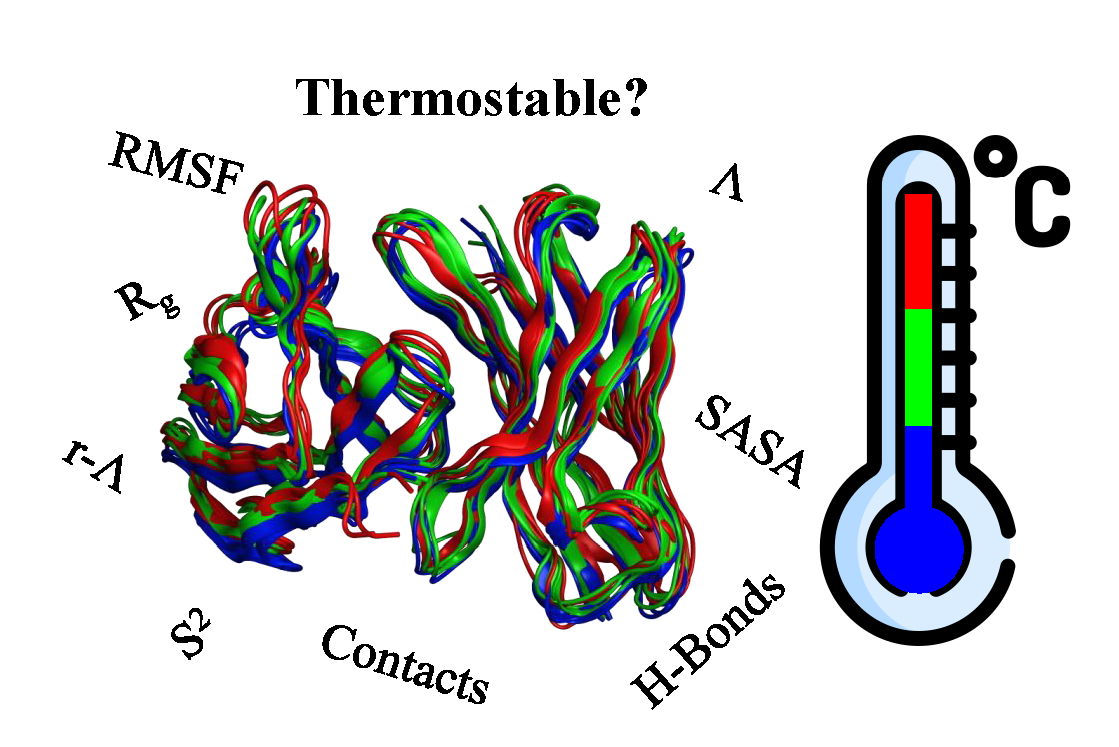
AbMelt: Learning Antibody Thermostability from Molecular Dynamics
Zachary Rollins, Postdoctoral Research Fellow,Merck Research Labs
Zachary Rollins, Postdoctoral Research Fellow,
Ionizable Amino Lipids Distribution and Effects on DSPC/cholesterol Membranes: Implications for Lipid Nanoparticle Structure
Sreyoshi Sur, Scientist,Moderna Inc.
Sreyoshi Sur, Scientist,
Next Generation of Multispecific Antibody Engineering
Fernando Garces, Director Protein Therapeutics,formerly of Gilead Sciences
Fernando Garces, Director Protein Therapeutics,
Closing Remarks
Social Hour with Posters