

Nous sommes heureux d'annoncer que la prochaine UGM et conférence européenne de CCG aura lieu du 21 au 24 mai à Lyon, en France. Il s'agit d'un événement de 4 jours, incluant des ateliers, des présentations scientifiques et une séance d'affiches scientifiques ainsi que des activités sociales comprenant des réceptions et un dîner de conférence. 2024 marque le 30e anniversaire de Chemical Computing Group. Rejoignez-nous pour célébrer cette occasion spéciale!
Si vous souhaitez faire une présentation scientifique à la conférence, veuillez contacter Steve Maginn à .
Registration Opens (Check-in/Badge pick-up)
Buffet Lunch
Peptide Modeling, Conformational Searching and Docking
Peptide Complex Preparation / Protein-Peptide Interaction Analysis / Surfaces and Maps / Peptide Sequence Optimization / Non-Natural Amino Acids / Conformational Searching / Peptide-Protein Docking / Protein-Peptide Interaction Fingerprints
Peptide Complex Preparation / Protein-Peptide Interaction Analysis / Surfaces and Maps / Peptide Sequence Optimization / Non-Natural Amino Acids / Conformational Searching / Peptide-Protein Docking / Protein-Peptide Interaction Fingerprints
Cheminformatics: Manage, Analyze, Model and Mine Molecular Data
MOE databases / Molecular Descriptors / Sorting and Coloring Plots / Clustering | Diverse Subset Selection / QSAR Modeling / Binary QSAR / Substructure Searching / Molecular Fingerprints / Similarity Searching
MOE databases / Molecular Descriptors / Sorting and Coloring Plots / Clustering | Diverse Subset Selection / QSAR Modeling / Binary QSAR / Substructure Searching / Molecular Fingerprints / Similarity Searching
Welcome Refreshments
Advanced Structure-Based Design
Protein-Ligand Interaction Analysis / Pharmacophore Modeling / Docking / Protein-Ligand Interaction Fingerprints (PLIF) / Fragment-based Design / Scaffold Replacement
Protein-Ligand Interaction Analysis / Pharmacophore Modeling / Docking / Protein-Ligand Interaction Fingerprints (PLIF) / Fragment-based Design / Scaffold Replacement
Antibody Modeling and Protein Engineering
Protein-Protein Interaction Analysis / Molecular Surfaces / Protein Patch Analysis / Protein Properties / Protein Engineering / Antibody Homology Modeling / Antibody Database / Developability Analysis
Protein-Protein Interaction Analysis / Molecular Surfaces / Protein Patch Analysis / Protein Properties / Protein Engineering / Antibody Homology Modeling / Antibody Database / Developability Analysis
Lunch Break
Small Molecule Virtual Screening
Virtual Screening Compound Libraries / Molecular Descriptors / QSAR/QSPR Modeling / Molecular Fingerprints / Pharmacophore Modeling / Filtering Compound Libraries / Pharmacophore-guided Docking / Template-based Docking / De novo Hit Expansion
Virtual Screening Compound Libraries / Molecular Descriptors / QSAR/QSPR Modeling / Molecular Fingerprints / Pharmacophore Modeling / Filtering Compound Libraries / Pharmacophore-guided Docking / Template-based Docking / De novo Hit Expansion
Biologics: Protein Alignments, Modeling and Docking
Protein Alignments and Superposition / Loop and Linker Modeling / Homology Modeling / Protein-Protein Docking / Epitope Analysis / Protein Properties / Protein Solubility Prediction / Protein Patches (2D and 3D) / Biologics QSAR/QSPR Modeling
Protein Alignments and Superposition / Loop and Linker Modeling / Homology Modeling / Protein-Protein Docking / Epitope Analysis / Protein Properties / Protein Solubility Prediction / Protein Patches (2D and 3D) / Biologics QSAR/QSPR Modeling
Opening Reception & Poster Session
Optional Walking Tour of Lyon
Welcome Refreshments
Opening Remarks
Exploring Multiple Approaches for Predicting Permeability of Cyclic Peptides
Claudia Beato, Principal Scientist,Evotec (Verona)
Claudia Beato, Principal Scientist,
Predicting Post-Translational Modifications through Structural In Silico Analysis
Elizabeth Rodriguez, Principal Scientist,UCB
Elizabeth Rodriguez, Principal Scientist,
Design of Site-Specific Antibody-Drug Conjugates through Computational Modelling
Elena de Orbe Izquierdo, Senior Scientist,AstraZeneca
Elena de Orbe Izquierdo, Senior Scientist,
Morning Break
Cryo-EM Illuminates Modern Drug Discovery: Unveiling the Molecular Landscape Where Other Methods Fall Short
Alexey Rak, Head of Bio Structure and Biophysics at Integrated Drug Discovery,Sanofi R&D (Vitry)
Alexey Rak, Head of Bio Structure and Biophysics at Integrated Drug Discovery,
Towards Machine Learning Approaches for Predicting Chemical Liability
Dilyana Dimova, Senior Data Scientist,Sanofi
Dilyana Dimova, Senior Data Scientist,
A Machine Learning Approach to Improving Antibody Developability
Paul MacDonald, Investigator,GSK
Paul MacDonald, Investigator,
Lunch Break
Enhancing the Potency and Stability of TCR-based Therapeutics through in silico Engineering
Tom Dixon, Senior Computational Structural Biologist,Etcembly Ltd.
Tom Dixon, Senior Computational Structural Biologist,
An All-inclusive Package for ADMET and Off-target Machine Learning
Anke Hackl, Data Computation Platform Lead,Bayer Pharma AG
Anke Hackl, Data Computation Platform Lead,
Extending & Testing Poisson-Boltzmann Electrostatics with MOE
Nicolas Foloppe, Principal Scientist,Vernalis
Nicolas Foloppe, Principal Scientist,
Afternoon Break
The Open Force Field Initiative: An Update on Our Progress and Plans
David Mobley, Professor, Pharmaceutical Sciences and Chemistry,University of California, Irvine
David Mobley, Professor, Pharmaceutical Sciences and Chemistry,
AmberEHT Forcefield: Novel Approaches to Small Molecule Parameterization
Paul Labute, President and CEO,Chemical Computing Group
Paul Labute, President and CEO,
Drinks Reception and Conference Dinner Cruise on the Rhône and Saône Rivers
Welcome Refreshments
Generative AI and Drug Design - Hallucinations and Reality
Richard Cooper, Head of Methods Development and Machine Learning,Oxford Drug Design
Richard Cooper, Head of Methods Development and Machine Learning,
Quantum Chemistry Calculations of Nitrosamine Activation and Deactivation Pathways for Carcinogenicity Risk Assessment
Andreas Göller, Principal Scientist and Bayer Senior Science Fellow,Bayer Pharma AG
Andreas Göller, Principal Scientist and Bayer Senior Science Fellow,
Deep Dive into Molecular Interactions: When Do They Translate into Measured in Vitro Binding/Activity?
Anna Karawajczyk, Director of Computational Chemistry,Selvita S.A.
Anna Karawajczyk, Director of Computational Chemistry,
Morning Break
Application of Absolute Binding Free Energy Calculations to Predict the Binding Modes and Affinities of Protein–Protein Inhibitors
Léa El Khoury, Group Leader, Computational Chemistry,Qubit Pharmaceuticals
Léa El Khoury, Group Leader, Computational Chemistry,
Modelling Protoporphyrinogen Oxidase
Michael Charlton, Fellow, Computational Chemistry,MoA Technology Ltd
Michael Charlton, Fellow, Computational Chemistry,
Probing the Psychedelic Structure-Activity Space
Chris Williams, Principal Scientist,Chemical Computing Group
Chris Williams, Principal Scientist,
Lunch Break
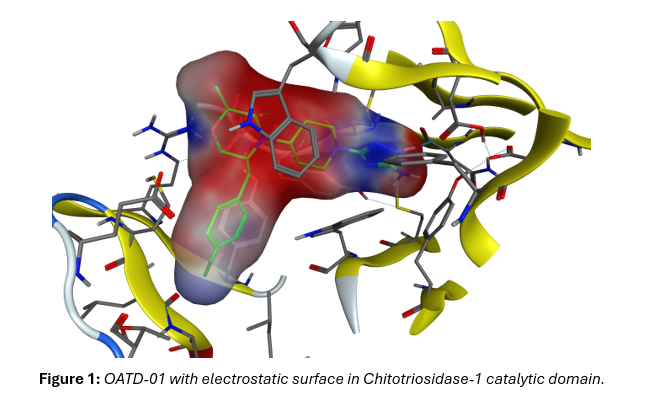
The Role of QM/MM MD Simulations in Revealing the Complexity of Chito-triosidase-1, Its Mode of Action and Inhibition by OATD-01
Mariusz Milewski, Cheminformatician,Molecure S.A.
Mariusz Milewski, Cheminformatician,
Closing Remarks
Closing Reception
La soumission d’affiches scientifiques est maintenant terminée.